一、写在前面
最近有粉丝提问,收到了如下的审稿人意见:

审稿人认为在单细胞测序过程中,利用findMarker通过Wilcox获得的差异基因虽然考虑到了不同组别细胞数量的不同,但是未能考虑到每组样本数量的不同。因此作者希望纳入样本水平的pseudo-bulk分析能够有助于确认两种条件下的差异基因。
首先我个人觉得审稿人自己的话有些矛盾,使用pseudo-bulk计算差异基因,岂不是无法考虑到不同组别中样本数量的差异?另外这有些吹毛求疵,在Seurat V5之前,作者甚至没有在包里集成pseudo-bulk的函数与算法(当然也可以自己提取矩阵计算)。难道能说作者发表的这几篇Cell和Nature给大家推荐的流程不好吗:
Hao, Hao, et al., Cell 2021 [Seurat v4]
Stuart, Butler, et al., Cell 2019 [Seurat v3]
Butler, et al., Nat Biotechnol 2018 [Seurat v2] Satija, Farrell, et al., Nat Biotechnol 2015 [Seurat v1]
再者说,scRNA-seq比Bulk RNA-Seq更加的稀疏,将前者模拟为后者参与差异计算,其实也没那么科学。当然,审稿人的观点也不是全无道理,若能够通过不同的算法得到相同的差异基因结果,的确有较高的说服力。
二、pseudo-bulk差异分析走起
测试文件可以自行下载:
链接:https://pan.baidu.com/s/12dEGTJy4DnQ7gH2mbxCf-A?pwd=7qfm
提取码:7qfm

2.1 数据载入
# 加载R包
library(Seurat)## 载入需要的程序包:SeuratObject## 载入需要的程序包:sp##
## 载入程序包:'SeuratObject'## The following objects are masked from 'package:base':
##
## intersect, t# 读取数据:
scRNA <- readRDS('test_data/T1D_scRNA.rds')# 这个数据包含24个样本:
unique(scRNA$sample)## [1] "D_503" "H_120" "H_630" "H_3060" "D_609" "H_727" "H_4579" "D_504"
## [9] "H_3128" "H_7108" "D_502" "D_497" "D_506" "H_409" "H_6625" "D_610"
## [17] "D_501" "D_500" "H_4119" "H_1334" "D_498" "H_2928" "D_644" "D_505"# 包含两个组别的数据:
DimPlot(scRNA,split.by = 'Group')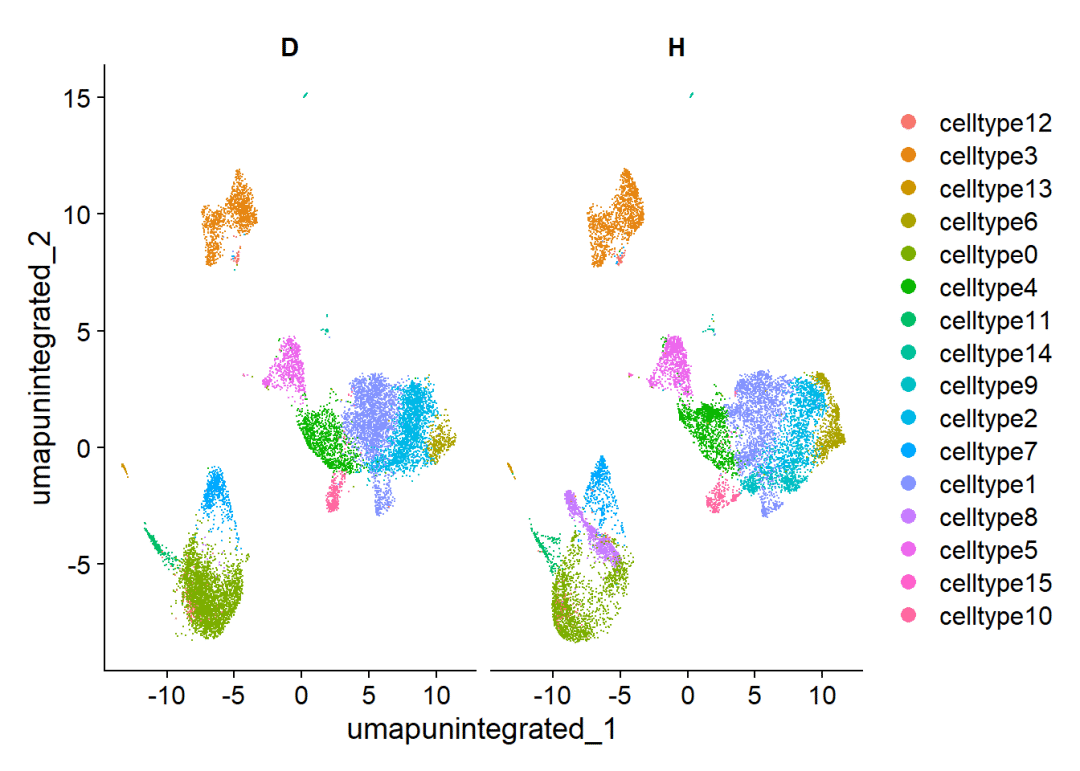
2.2 差异计算
(1) pseudo-bulk差异计算
### 生成拟bulk 数据 ###
bulk <- AggregateExpression(scRNA, return.seurat = T, slot = "counts", assays = "RNA",group.by = c("cell_type", "sample", "Group")# 分别填写细胞类型、样本变量、分组变量的slot名称)## Names of identity class contain underscores ('_'), replacing with dashes ('-')
## Centering and scaling data matrix
##
## This message is displayed once every 8 hours.# 生成的是一个新的Seurat对象
bulk## An object of class Seurat
## 41056 features across 345 samples within 1 assay
## Active assay: RNA (41056 features, 0 variable features)
## 3 layers present: counts, data, scale.data我们可以像普通scRNA-seq的Seurat对象一样,利用FindMarkers()进行差异分析,我们这里用celltype10做演示。
# 取出celltype10对应的对象:
ct10.bulk <- subset(bulk, cell_type == "celltype10")
# 改变默认分类变量:
Idents(ct10.bulk) <- "Group"# 下面的额计算依赖DESeq2,做过Bulk RNA-Seq的同学都知道:
if(!require(DESeq2))BiocManager::install('DESeq2')## 载入需要的程序包:DESeq2## 载入需要的程序包:S4Vectors## 载入需要的程序包:stats4## 载入需要的程序包:BiocGenerics##
## 载入程序包:'BiocGenerics'## The following object is masked from 'package:SeuratObject':
##
## intersect## The following objects are masked from 'package:stats':
##
## IQR, mad, sd, var, xtabs## The following objects are masked from 'package:base':
##
## anyDuplicated, aperm, append, as.data.frame, basename, cbind,
## colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
## get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply,
## match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
## Position, rank, rbind, Reduce, rownames, sapply, setdiff, table,
## tapply, union, unique, unsplit, which.max, which.min##
## 载入程序包:'S4Vectors'## The following object is masked from 'package:utils':
##
## findMatches## The following objects are masked from 'package:base':
##
## expand.grid, I, unname## 载入需要的程序包:IRanges##
## 载入程序包:'IRanges'## The following object is masked from 'package:sp':
##
## %over%## The following object is masked from 'package:grDevices':
##
## windows## 载入需要的程序包:GenomicRanges## 载入需要的程序包:GenomeInfoDb## 载入需要的程序包:SummarizedExperiment## 载入需要的程序包:MatrixGenerics## 载入需要的程序包:matrixStats##
## 载入程序包:'MatrixGenerics'## The following objects are masked from 'package:matrixStats':
##
## colAlls, colAnyNAs, colAnys, colAvgsPerRowSet, colCollapse,
## colCounts, colCummaxs, colCummins, colCumprods, colCumsums,
## colDiffs, colIQRDiffs, colIQRs, colLogSumExps, colMadDiffs,
## colMads, colMaxs, colMeans2, colMedians, colMins, colOrderStats,
## colProds, colQuantiles, colRanges, colRanks, colSdDiffs, colSds,
## colSums2, colTabulates, colVarDiffs, colVars, colWeightedMads,
## colWeightedMeans, colWeightedMedians, colWeightedSds,
## colWeightedVars, rowAlls, rowAnyNAs, rowAnys, rowAvgsPerColSet,
## rowCollapse, rowCounts, rowCummaxs, rowCummins, rowCumprods,
## rowCumsums, rowDiffs, rowIQRDiffs, rowIQRs, rowLogSumExps,
## rowMadDiffs, rowMads, rowMaxs, rowMeans2, rowMedians, rowMins,
## rowOrderStats, rowProds, rowQuantiles, rowRanges, rowRanks,
## rowSdDiffs, rowSds, rowSums2, rowTabulates, rowVarDiffs, rowVars,
## rowWeightedMads, rowWeightedMeans, rowWeightedMedians,
## rowWeightedSds, rowWeightedVars## 载入需要的程序包:Biobase## Welcome to Bioconductor
##
## Vignettes contain introductory material; view with
## 'browseVignettes()'. To cite Bioconductor, see
## 'citation("Biobase")', and for packages 'citation("pkgname")'.##
## 载入程序包:'Biobase'## The following object is masked from 'package:MatrixGenerics':
##
## rowMedians## The following objects are masked from 'package:matrixStats':
##
## anyMissing, rowMedians##
## 载入程序包:'SummarizedExperiment'## The following object is masked from 'package:Seurat':
##
## Assays## The following object is masked from 'package:SeuratObject':
##
## Assays# 差异计算:
bulk_deg <- FindMarkers(ct10.bulk, ident.1 = "D", ident.2 = "H", # 这样算出来的Fold Change就是D/Hslot = "counts", test.use = "DESeq2",# 这里可以选择其它算法verbose = F# 关闭进度提示)## converting counts to integer mode## gene-wise dispersion estimates## mean-dispersion relationship## final dispersion estimateshead(bulk_deg)# 看一下差异列表## p_val avg_log2FC pct.1 pct.2 p_val_adj
## ENSG00000047346 1.900955e-05 -1.1201537 0.917 1.000 0.780456
## ENSG00000168685 7.606182e-05 -0.5817112 1.000 1.000 1.000000
## ENSG00000131759 9.696591e-05 -2.1668759 0.667 1.000 1.000000
## ENSG00000166750 2.136829e-04 -1.5473545 0.750 0.917 1.000000
## ENSG00000163947 3.127113e-04 -0.9136378 0.917 1.000 1.000000
## ENSG00000239713 4.090397e-04 -2.2337008 0.250 0.917 1.000000如何写循环计算所有细胞类型的差异基因,就留在这里当习题啦。
(2)细胞水平的差异计算
# 整理分组变量:
scRNA$CT_Group <- paste(scRNA$cell_type,scRNA$Group,sep = '_')# 查看新的分组变量:
unique(scRNA$CT_Group)## [1] "celltype12_D" "celltype3_H" "celltype13_H" "celltype6_H" "celltype0_D"
## [6] "celltype12_H" "celltype4_H" "celltype11_D" "celltype14_H" "celltype9_H"
## [11] "celltype11_H" "celltype2_H" "celltype0_H" "celltype7_H" "celltype14_D"
## [16] "celltype1_D" "celltype4_D" "celltype1_H" "celltype8_H" "celltype3_D"
## [21] "celltype13_D" "celltype8_D" "celltype7_D" "celltype5_H" "celltype6_D"
## [26] "celltype15_H" "celltype2_D" "celltype5_D" "celltype10_H" "celltype9_D"
## [31] "celltype10_D" "celltype15_D"# 差异计算:
cell_deg <- FindMarkers(scRNA,ident.1 = 'celltype10_D',ident.2 = 'celltype10_H' ,group.by = 'CT_Group')# 同样得到的是celltype10在D组 vs H组的结果## For a (much!) faster implementation of the Wilcoxon Rank Sum Test,
## (default method for FindMarkers) please install the presto package
## --------------------------------------------
## install.packages('devtools')
## devtools::install_github('immunogenomics/presto')
## --------------------------------------------
## After installation of presto, Seurat will automatically use the more
## efficient implementation (no further action necessary).
## This message will be shown once per session(3)两种算法的对比
library(dplyr)##
## 载入程序包:'dplyr'## The following object is masked from 'package:Biobase':
##
## combine## The following object is masked from 'package:matrixStats':
##
## count## The following objects are masked from 'package:GenomicRanges':
##
## intersect, setdiff, union## The following object is masked from 'package:GenomeInfoDb':
##
## intersect## The following objects are masked from 'package:IRanges':
##
## collapse, desc, intersect, setdiff, slice, union## The following objects are masked from 'package:S4Vectors':
##
## first, intersect, rename, setdiff, setequal, union## The following objects are masked from 'package:BiocGenerics':
##
## combine, intersect, setdiff, union## The following objects are masked from 'package:stats':
##
## filter, lag## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union# 先看一下两种算法的显著差异基因数量
bulk_sig <- filter(bulk_deg,p_val < 0.05)
nrow(bulk_sig)## [1] 308cell_sig <- filter(cell_deg,p_val < 0.05)
nrow(cell_sig)## [1] 1494可以看出,pseudo-bulk得到的差异基因数量要少很多,画一个韦恩图看看二者交集
if(!require(VennDiagram))install.packages("VennDiagram")## 载入需要的程序包:VennDiagram## 载入需要的程序包:grid## 载入需要的程序包:futile.loggervenn.plot <- venn.diagram(x = list(Bulk = rownames(bulk_sig), Cell = rownames(cell_sig)
),category.names = c("Bulk DEG", "Single-Cell DEG"),filename = NULL,output = TRUE,main = "Venn Diagram of Significant Genes"
)
grid.draw(venn.plot)
可以看出包含关系还是挺明显的,那我们再用交集基因的avg_log2FC做一个线性回归看看两次差异分析的相关性如何:
# 获得两次差异分析共同出现的基因:
inter_gene <- intersect(rownames(bulk_sig),rownames(cell_sig))
# 取出avg_log2FC整理为数据框
data4plot <- data.frame(Bulk = bulk_sig[inter_gene,'avg_log2FC'],Cell = cell_sig[inter_gene,'avg_log2FC'] )# 线性回归分析:
lm.model <- lm(Bulk ~ Cell,data = data4plot)
summary(lm.model)#看一下统计学参数##
## Call:
## lm(formula = Bulk ~ Cell, data = data4plot)
##
## Residuals:
## Min 1Q Median 3Q Max
## -1.01613 -0.24964 -0.04723 0.17148 2.19351
##
## Coefficients:
## Estimate Std. Error t value Pr(>|t|)
## (Intercept) -0.17249 0.02757 -6.257 1.58e-09 ***
## Cell 0.70081 0.01959 35.767 < 2e-16 ***
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## Residual standard error: 0.4107 on 263 degrees of freedom
## Multiple R-squared: 0.8295, Adjusted R-squared: 0.8288
## F-statistic: 1279 on 1 and 263 DF, p-value: < 2.2e-16mypara <- coefficients(lm.model)#得到截距和斜率
a <- mypara[2]#斜率
b <- mypara[1]#截距
a <- round(a,2)#取两位有效数字
b <- round(b,2)library(ggplot2)
library(ggpubr)##
## 载入程序包:'ggpubr'## The following object is masked from 'package:VennDiagram':
##
## rotate# 来个散点图吧~
lmplot <- ggplot( data4plot,aes(x=Bulk, y=Cell))+geom_point(color="black")+stat_smooth(method="lm",se=TRUE)+stat_cor(data=data4plot, method = "pearson")+#加上置信区间、R值、P值ggtitle(label = paste(": y = ", a, " * x + ", b, sep = ""))+geom_rug()+#加上线性回归方程labs(x='Bulk DEG',y= 'single-cell DEG')lmplot## `geom_smooth()` using formula = 'y ~ x'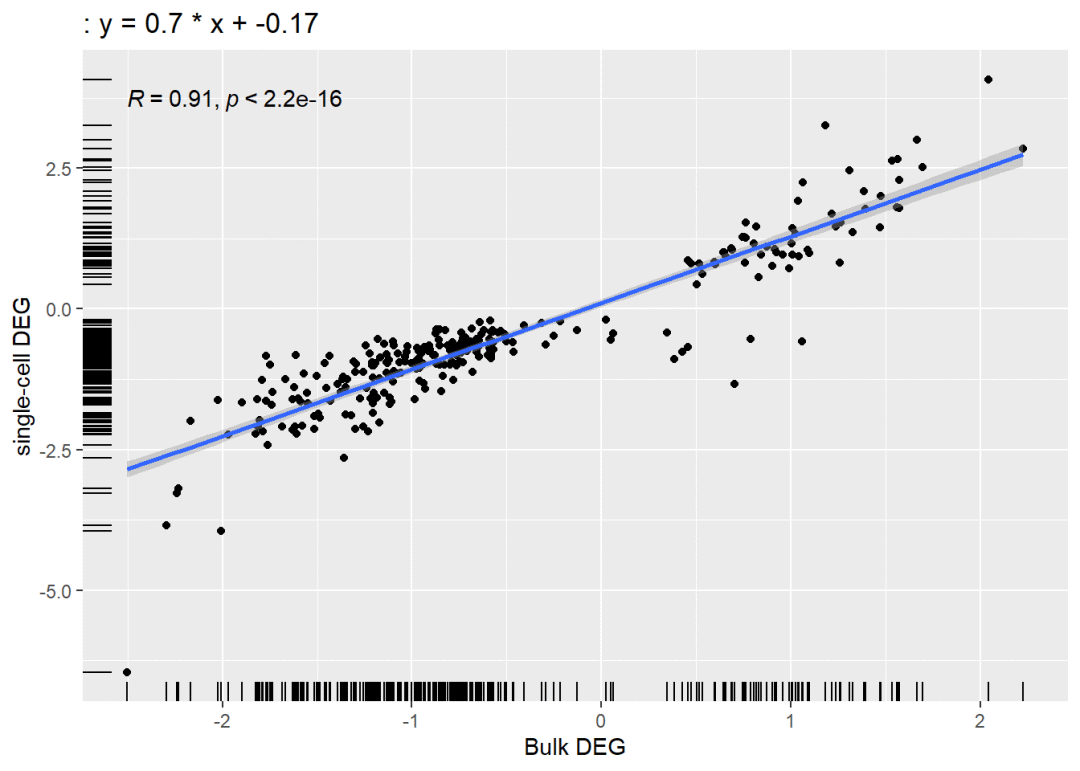
R=0.91,那么R^2就是0.83,可以看出二者的相关性还是不错的,就看能不能过审稿人这关啦。
环境信息
sessionInfo()## R version 4.4.1 (2024-06-14 ucrt)
## Platform: x86_64-w64-mingw32/x64
## Running under: Windows 11 x64 (build 22631)
##
## Matrix products: default
##
##
## locale:
## [1] LC_COLLATE=Chinese (Simplified)_China.utf8
## [2] LC_CTYPE=Chinese (Simplified)_China.utf8
## [3] LC_MONETARY=Chinese (Simplified)_China.utf8
## [4] LC_NUMERIC=C
## [5] LC_TIME=Chinese (Simplified)_China.utf8
##
## time zone: Asia/Shanghai
## tzcode source: internal
##
## attached base packages:
## [1] grid stats4 stats graphics grDevices utils datasets
## [8] methods base
##
## other attached packages:
## [1] ggpubr_0.6.0 ggplot2_3.5.1
## [3] VennDiagram_1.7.3 futile.logger_1.4.3
## [5] dplyr_1.1.4 DESeq2_1.44.0
## [7] SummarizedExperiment_1.34.0 Biobase_2.64.0
## [9] MatrixGenerics_1.16.0 matrixStats_1.4.1
## [11] GenomicRanges_1.56.1 GenomeInfoDb_1.40.1
## [13] IRanges_2.38.1 S4Vectors_0.42.1
## [15] BiocGenerics_0.50.0 Seurat_5.1.0
## [17] SeuratObject_5.0.2 sp_2.1-4
##
## loaded via a namespace (and not attached):
## [1] RcppAnnoy_0.0.22 splines_4.4.1 later_1.3.2
## [4] tibble_3.2.1 polyclip_1.10-7 fastDummies_1.7.4
## [7] lifecycle_1.0.4 rstatix_0.7.2 globals_0.16.3
## [10] lattice_0.22-6 MASS_7.3-60.2 backports_1.5.0
## [13] magrittr_2.0.3 plotly_4.10.4 sass_0.4.9
## [16] rmarkdown_2.28 jquerylib_0.1.4 yaml_2.3.10
## [19] httpuv_1.6.15 sctransform_0.4.1 spam_2.10-0
## [22] spatstat.sparse_3.1-0 reticulate_1.39.0 cowplot_1.1.3
## [25] pbapply_1.7-2 RColorBrewer_1.1-3 abind_1.4-5
## [28] zlibbioc_1.50.0 Rtsne_0.17 purrr_1.0.2
## [31] GenomeInfoDbData_1.2.12 ggrepel_0.9.6 irlba_2.3.5.1
## [34] listenv_0.9.1 spatstat.utils_3.1-0 openintro_2.5.0
## [37] airports_0.1.0 goftest_1.2-3 RSpectra_0.16-2
## [40] spatstat.random_3.3-1 fitdistrplus_1.2-1 parallelly_1.38.0
## [43] leiden_0.4.3.1 codetools_0.2-20 DelayedArray_0.30.1
## [46] tidyselect_1.2.1 UCSC.utils_1.0.0 farver_2.1.2
## [49] spatstat.explore_3.3-2 jsonlite_1.8.8 progressr_0.14.0
## [52] ggridges_0.5.6 survival_3.6-4 tools_4.4.1
## [55] ica_1.0-3 Rcpp_1.0.13 glue_1.7.0
## [58] gridExtra_2.3 SparseArray_1.4.8 mgcv_1.9-1
## [61] xfun_0.47 withr_3.0.1 formatR_1.14
## [64] fastmap_1.2.0 fansi_1.0.6 digest_0.6.37
## [67] R6_2.5.1 mime_0.12 colorspace_2.1-1
## [70] scattermore_1.2 tensor_1.5 spatstat.data_3.1-2
## [73] utf8_1.2.4 tidyr_1.3.1 generics_0.1.3
## [76] data.table_1.16.0 usdata_0.3.1 httr_1.4.7
## [79] htmlwidgets_1.6.4 S4Arrays_1.4.1 uwot_0.2.2
## [82] pkgconfig_2.0.3 gtable_0.3.5 lmtest_0.9-40
## [85] XVector_0.44.0 htmltools_0.5.8.1 carData_3.0-5
## [88] dotCall64_1.1-1 scales_1.3.0 png_0.1-8
## [91] spatstat.univar_3.0-1 knitr_1.48 lambda.r_1.2.4
## [94] rstudioapi_0.16.0 tzdb_0.4.0 reshape2_1.4.4
## [97] nlme_3.1-164 cachem_1.1.0 zoo_1.8-12
## [100] stringr_1.5.1 KernSmooth_2.23-24 parallel_4.4.1
## [103] miniUI_0.1.1.1 pillar_1.9.0 vctrs_0.6.5
## [106] RANN_2.6.2 promises_1.3.0 car_3.1-2
## [109] xtable_1.8-4 cluster_2.1.6 evaluate_0.24.0
## [112] readr_2.1.5 cli_3.6.3 locfit_1.5-9.10
## [115] compiler_4.4.1 futile.options_1.0.1 rlang_1.1.4
## [118] crayon_1.5.3 future.apply_1.11.2 ggsignif_0.6.4
## [121] labeling_0.4.3 plyr_1.8.9 stringi_1.8.4
## [124] viridisLite_0.4.2 deldir_2.0-4 BiocParallel_1.38.0
## [127] munsell_0.5.1 lazyeval_0.2.2 spatstat.geom_3.3-2
## [130] Matrix_1.7-0 RcppHNSW_0.6.0 hms_1.1.3
## [133] patchwork_1.2.0 future_1.34.0 shiny_1.9.1
## [136] highr_0.11 ROCR_1.0-11 broom_1.0.6
## [139] igraph_2.0.3 bslib_0.8.0 cherryblossom_0.1.0欢迎致谢
如果以上内容对你有帮助,欢迎在文章的Acknowledgement中加上这一段,联系客服微信可以发放奖励:
Since Biomamba and his wechat public account team produce bioinformatics tutorials and share code with annotation, we thank Biomamba for their guidance in bioinformatics and data analysis for the current study. 欢迎在发文/毕业时向我们分享你的喜悦~






