前言:
在前面的蛋白互作组学系列文章中(蛋白互作组学系列丨(七)蛋白互作结构域的预测分析),我们曾向大家分享过如何使用ZDOCK进行蛋白相互作用结构域预测与分析。这一期我们将向大家展示如何使用Pymol(https://pymol.org/2/)对蛋白互作结构域进行可视化分析。
Pymol是一个很强大的蛋白结构分析可视化软件。这一期的蛋白相互作用结构域可视化分析与后续的小分子-蛋白对接专题都会使用到Pymol这个软件。其操作界面和各部分的功能如图1所示。今天我们展示的分析流程需要先用Pymol对蛋白结构进行预处理,然后用ZDOCK进行分子对接,再用Pymol将对接结果进行可视化。
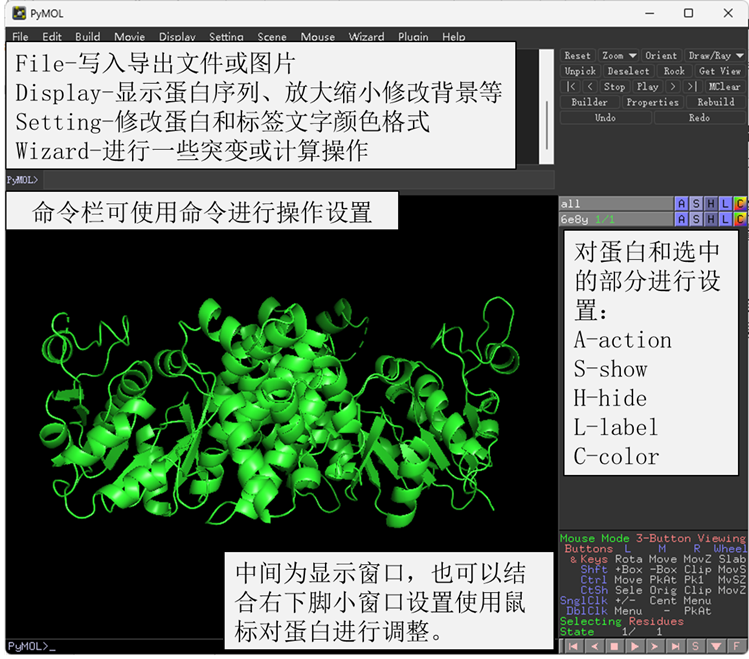
图 1 Pymol使用简介
一、蛋白结构下载与预处理
以已知的相互作用蛋白ENOA(P06733)与GAPDH(P04406)为例,先从PDB数据库(https://www.rcsb.org/)中下载他们的PDB结构,ID号分别为2psn与1u8f。因为PDB结构可能包含水分子或其他离子或配体,甚至是多聚体结构,因此在对接前需要使用Pymol对蛋白结构进行预处理。
以ENO1为例,其结构为四聚体,在Pymol中打开其结构文件,点击右下角S按钮显示其序列,拖动左上角3处划钮选择A链之外的序列和O(水分子),右键选择remove,即可只保留ENO1单体(图2-3)。选择File-Export molecule将ENO1单体保存为新的PDB文件(图4)。以同样的方法处理GAPDH结构。
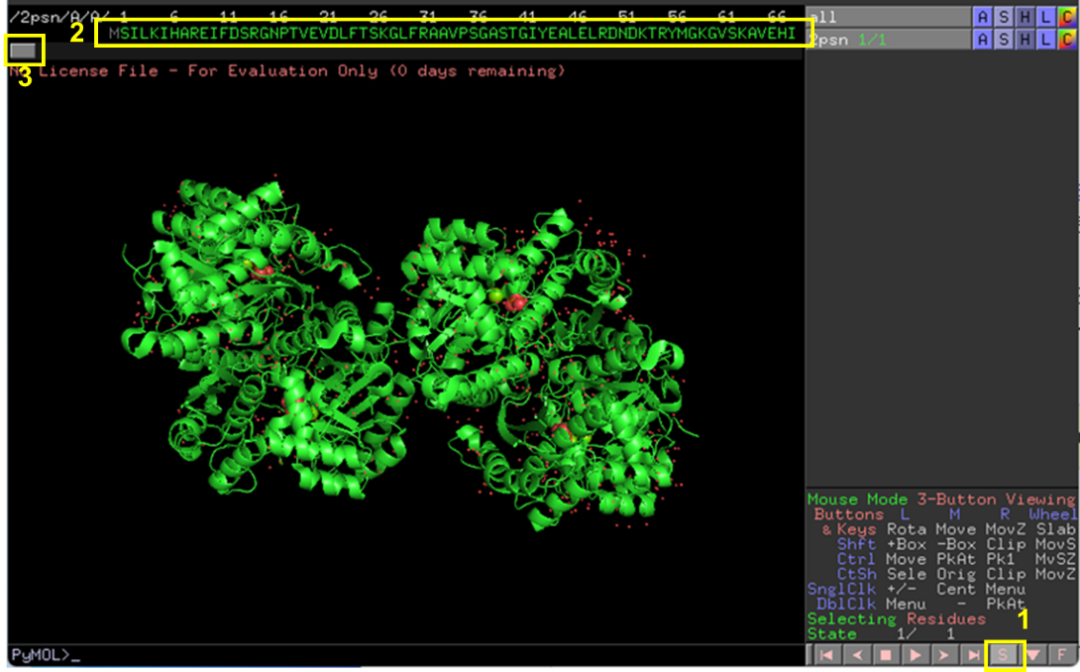
图 2 提取蛋白单体结构
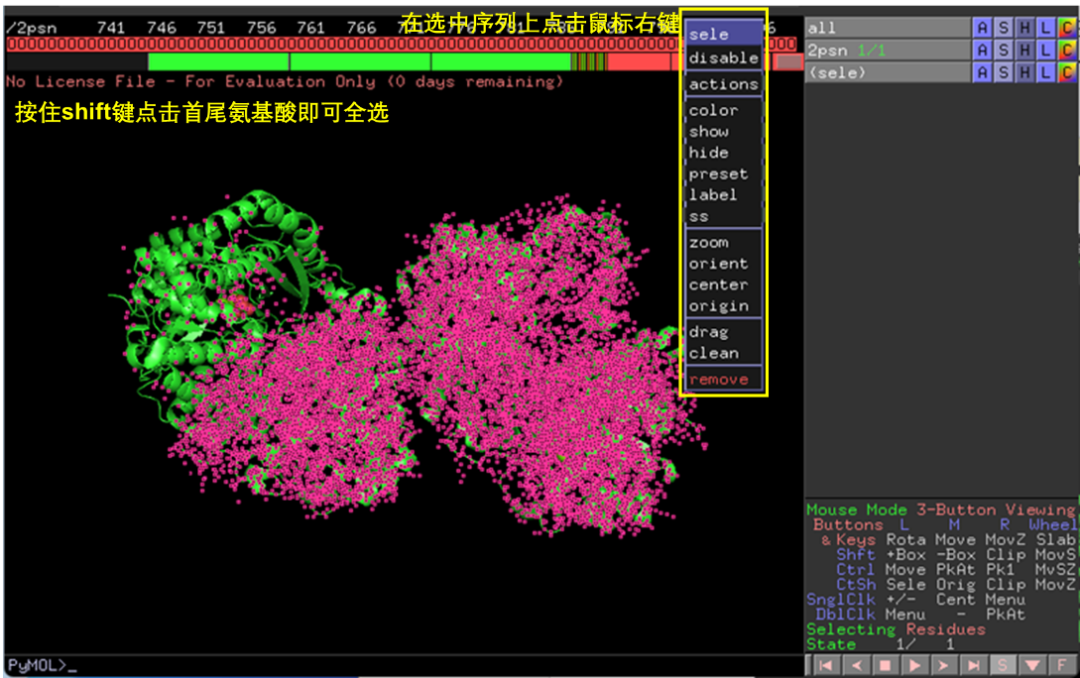
图 3 提取蛋白单体结构
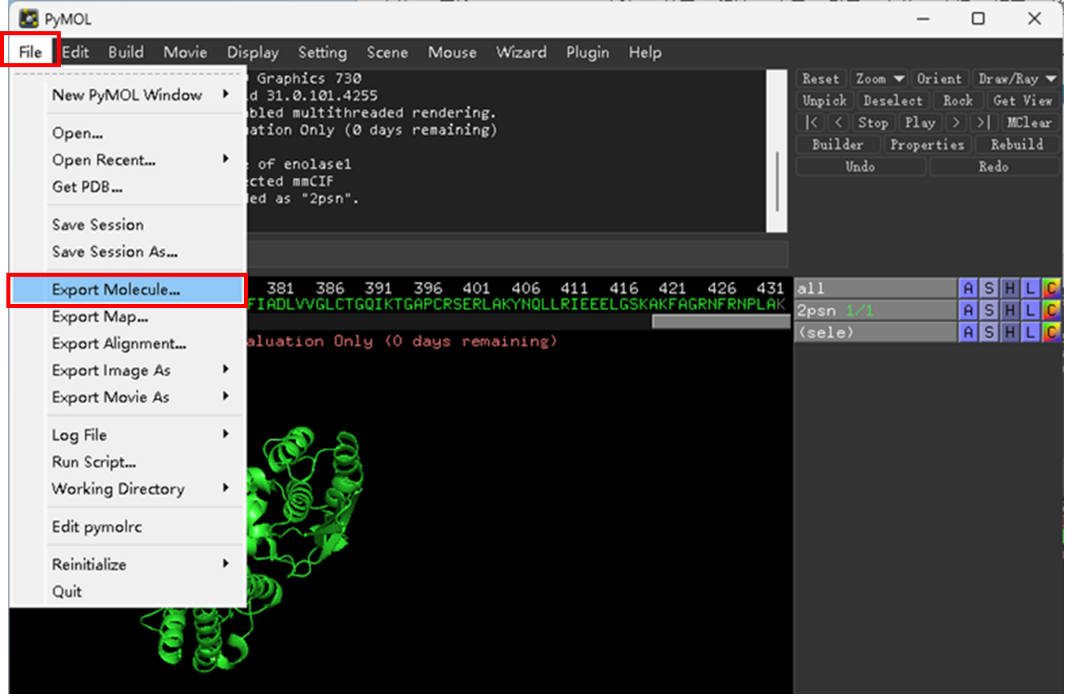
图 4 保存蛋白单体结构文件
二、ZDOCK蛋白相互作用分析
ZDOCK网址:ZDOCK Server: An automatic protein docking server (umassmed.edu)。具体的操作步骤我们在之前的文章《蛋白互作组学系列丨(七)蛋白互作结构域的预测分析》中介绍过。
图 5 ZDOCK在线服务界面
三、可视化对接结果
ZDOCK分析完成后,从邮箱中下载对接结果。可以使用前期所讲的PDBePISA对蛋白相互作用面进行分析。这一期我们将使用Pymol对蛋白的相互作用界面进行可视化分析。
1)分析对接结构中蛋白间相互作用
使用Pymol打开排名第一的相互作用结构。点击右上角C-by chain将两条链(两个蛋白)标上不同的颜色(图6)。点击A-find-polar contacts-between chains展示两个蛋白间的极性相互作用,这里主要是氢键(图7)。
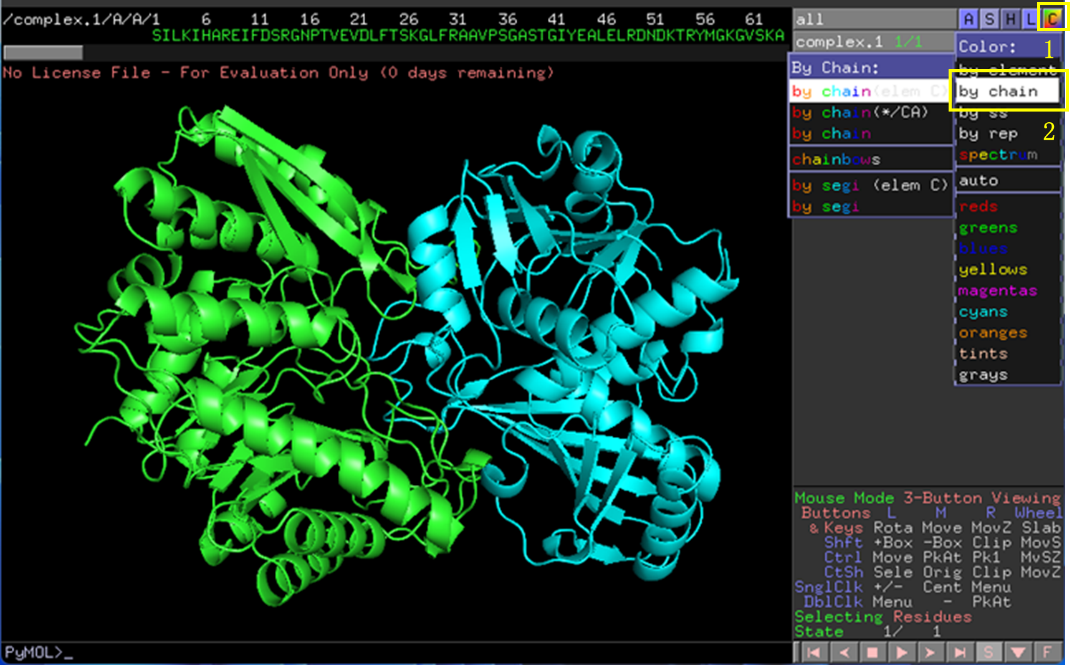
图 6 对接结构分析
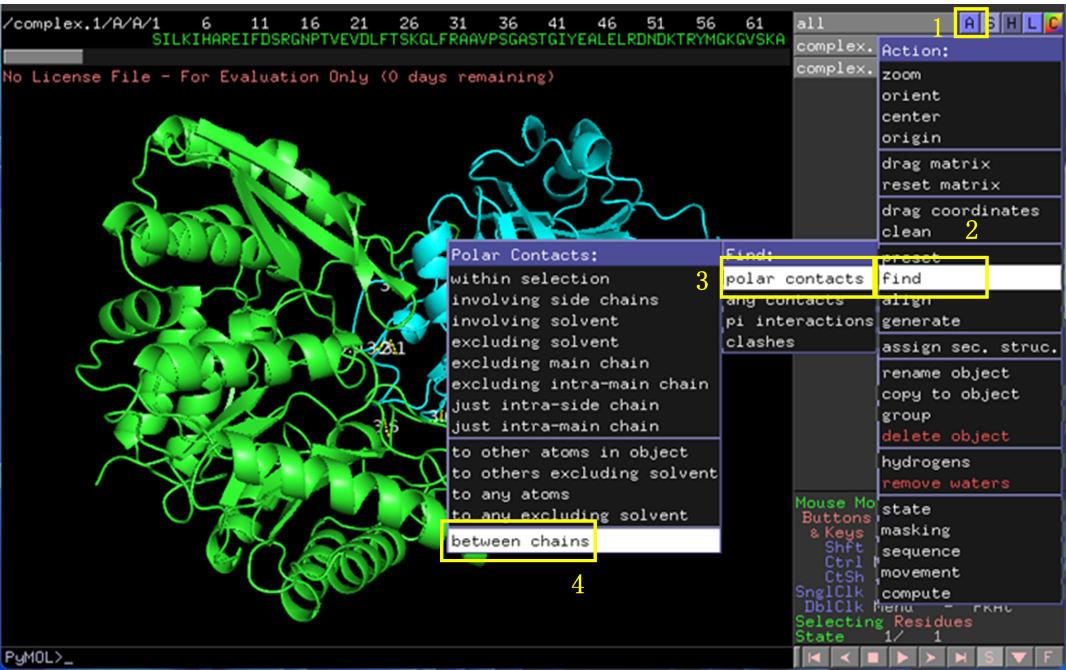
图 7 蛋白相互作用分析
氢键上的数字为键两端原子间的距离(单位Å),点击H-labels可隐藏数字(方便观察),也可以点击S-labels重新展示(图8)。
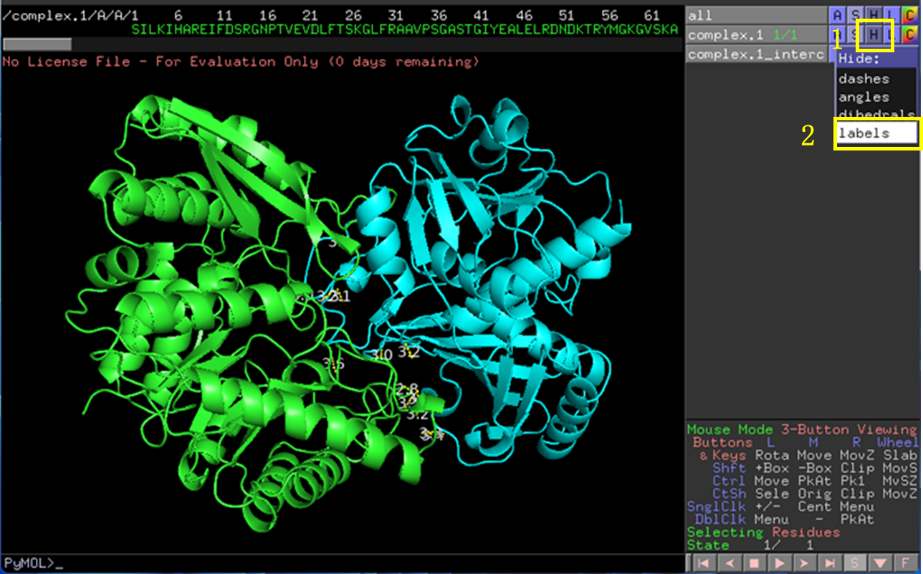
图 8 相互作用距离Lable展示与隐藏
2)展示相互作用氨基酸残基
通过鼠标点击可选中氢键两端的氨基酸残基,点击S-lines可展示相互作用氨基酸残基的结构(图9)。
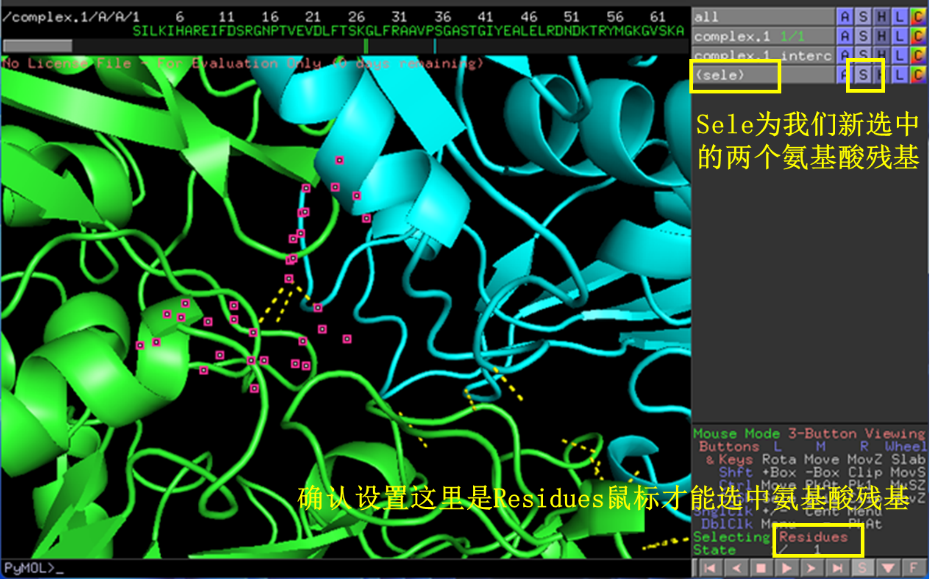
图 9 寻找展示相互作用氨基酸残基
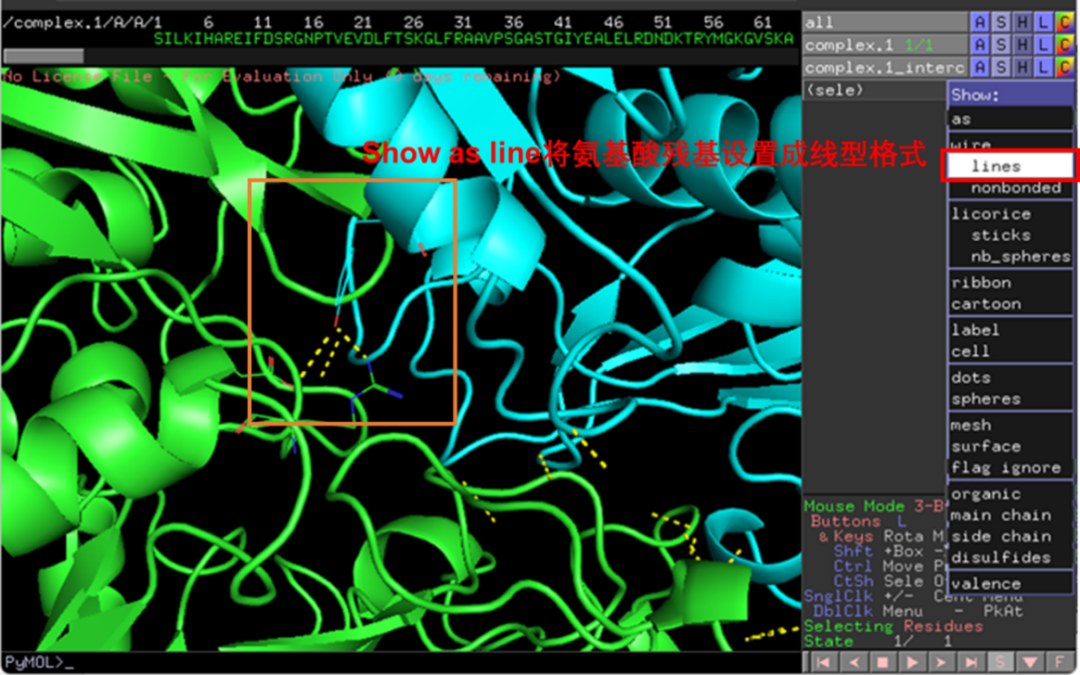
图 10 展示相互作用氨基酸残基线状结构
点击L-residues可展示相互作用氨基酸的名称,同样此处也可以通过H-labels隐藏(图11)。
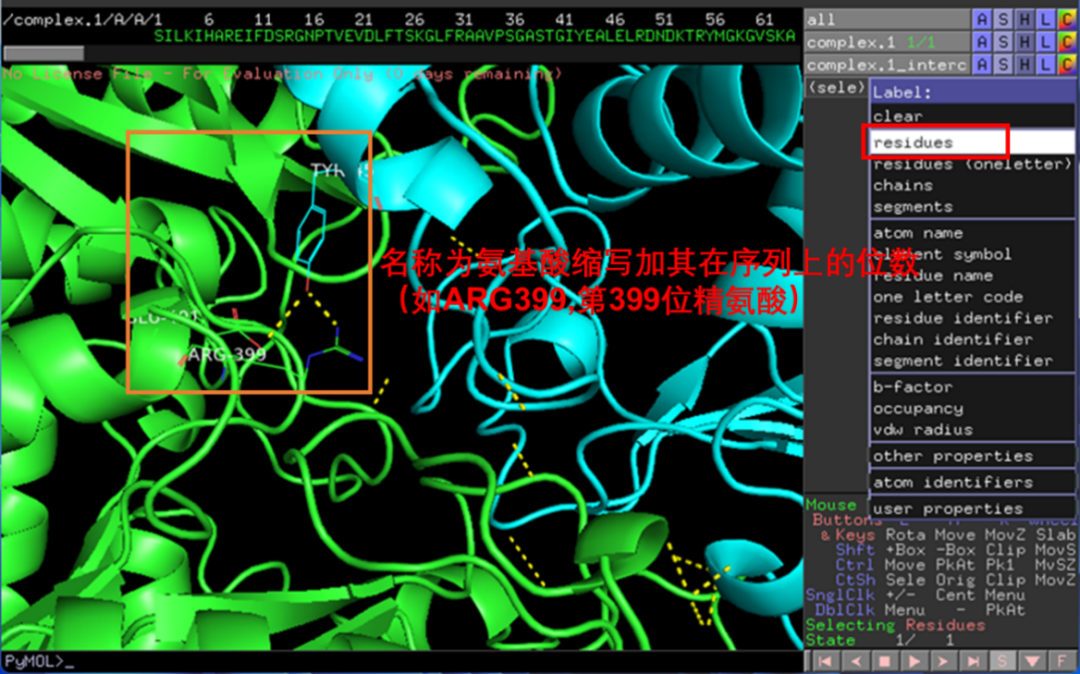
图 11 氨基酸名称展示与隐藏
以同样的步骤将所有的相互作用氨基酸残基都转换成线状形式后,即可得到如下图所示的蛋白相互作用界面。通过图片,我们可以观察到ENOA与GAPDH的相互作用依赖于其相互作用界面的氨基酸残基,总共涉及到ENOA蛋白上的10个氨基酸残基与GAPDH上的9个氨基酸残基间的相互作用(如GAPHD的第45位TYR45与ENOA的第399位ARG399和第401位GLU401相互作用)(图12)。

图 12 蛋白相互作用分析
3)导出蛋白相互作用结构图
点击Display-background可以设置背景颜色,于右上角的Draw/Ray处调整图片分辨率并导出图片。最终的效果如图13所示。
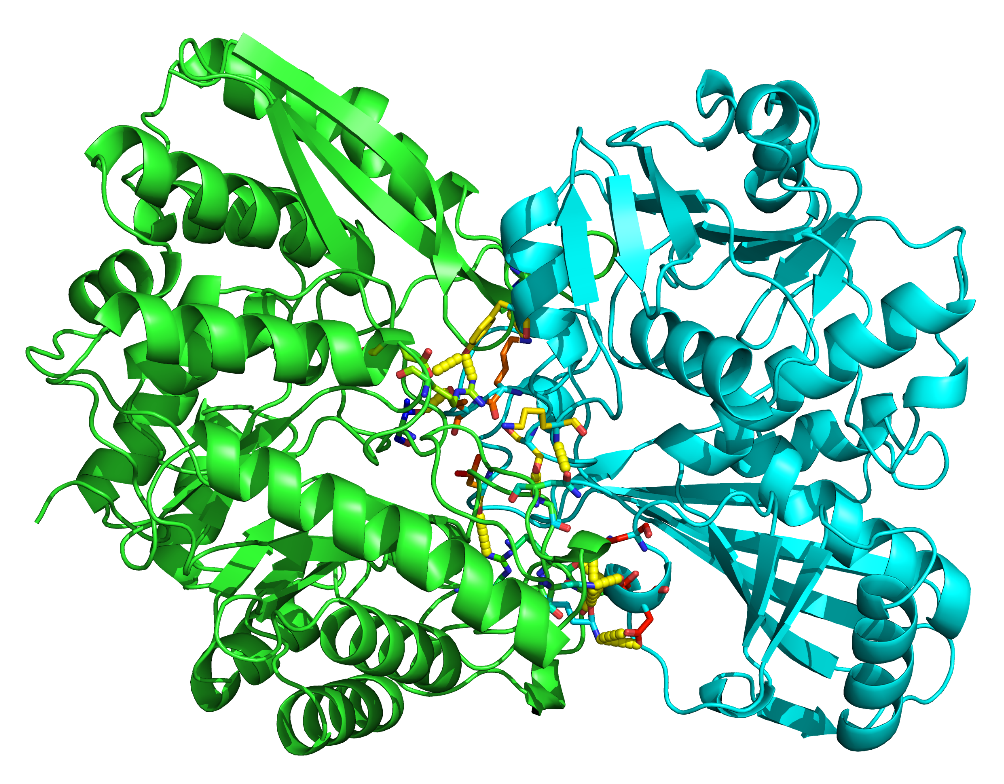
图 13 蛋白互作结构可视化分析结果图
以上就是我们今天的分享内容,在实际使用中,我们可能还会遇到以下疑问:
1)Q:在进行蛋白相互作用域可视化分析时,蛋白结构应该选择来源于实验的PDB结构还是来源于AlphaFold的预测结构?
A:如果有完整序列的PDB结构,优先使用PDB结构,如果没有,则可以使用AlphaFold结构。另外,因为PDB结构可能包含水分子或其他离子或配体,还有的是多聚体结构,因此在对接前需要对蛋白结构进行前处理。而AlphaFold结构是纯净的蛋白单体,可以直接使用。
2)Q:蛋白相互作用结构域预测(蛋白对接)的可信度如何?
A:相比而言,小分子-蛋白对接的可靠性更高,而蛋白-蛋白对接因为蛋白结构较大,而且会有结构柔性变化,因此较难预测。这里所使用ZDOCK是基于FFTs算法的刚性对接,使用较为广泛,对接结果可靠性较高。此外,感兴趣的老师可以关注一下CAPRI-全球蛋白复合物结构预测比赛,里面还会分享一些在蛋白相互作用预测中表现好的软件工具。因为蛋白-蛋白对接的计算量较大,在这里推荐使用在线服务器平台。


)
)

