这里写自定义目录标题
- ASE 介绍
- ASE 的安装
- 文件格式的转化
- 命令行操作
- 图形界面操作
- Python代码操作[2]
- 支持的文件转化格式
- ASE(三):图形界面查看/建立/修改结构
- 图形界面召唤
- 查看结构
- python代码查看/建立结构
- 读写、查看结构
- 建立结构
- 分子
- 周期性体系
https://nb.bohrium.dp.tech/detail/7217528884?utm_source=csdn
https://zhuanlan.zhihu.com/p/446086740?utm_id=0
https://blog.csdn.net/qq_40481843/article/details/128170814
pip install --upgrade ase
- read(),write()函数,作用分别是:将结构信息读取为atom object,将atom object写入文件;
- atom_object.get_positions()函数,可以获取atom object中所有的原子坐标信息,数据形式为列表;
- atom.index,atom.symbol,可以获取atom object中某种元素所对应所有原子索引
del atom_object[atom_index] ,删除atom object中某个原子
ASE 介绍
ASE (Atomic Simulation Environment) 是计算化学/计算材料领域的一个很好用的工具,能够用于建模、文件格式转化、计算结果分析与可视化,它内置了结构优化、过渡态计算、分子动力学模拟等一些常用的计算方法。同时,ASE有很多常用计算软件的接口 (calculators),用于生成输入文件,批量处理计算与运行。
它是一个Python库,同时也有图形界面 (GUI)和命令行工具,非常容易上手。官网的Tutorials有很多计算的示例代码,对计算小白超级友好。
GUI:https://wiki.fysik.dtu.dk/ase/ase/gui/gui.html#index-0
命令行工具:https://wiki.fysik.dtu.dk/ase/cmdline.html
示例:https://wiki.fysik.dtu.dk/ase/tutorials/tutorials.html

从结构的初始建模,输入文件中参数设置,计算任务提交,到结果可视化与分析的一个完整计算流程,都可以通过ASE完成,更简单来说,通过一个Python文件就可以完成。
虽然针对不同的计算软件已有许多计算脚本,比如VASPKIT[6],Gaussian的一些脚本[7]等,但是这些脚本往往只适用于某一个特定的软件。如果在研究中需要使用多个软件进行计算,需要批量生成或处理文件,ASE是一个不错的选择。
ASE 的安装
https://wiki.fysik.dtu.dk/ase/install.html
无论是Windows, Linux还是Mac OS X系统的安装,官网都有详细的介绍。安装不难,一步步跟着教程做即可。
装完ASE以后记得设置环境变量!
(这样可以使用命令行方式快速使用 ase)
文件格式的转化
使用Material Studio建模,用VASP做计算时通常需要将构建的xsd或cif文件转化为POSCAR文件进行输入。
有许多脚本/软件可以做到这一点,比如:
- 用VESTA打开cif文件,另存为.vasp文件,修改名称为POSCAR。(VESTA的好处是能够选择使用direct坐标还是cartesian坐标)
- 使用VASPKIT的105或106的cif2pos.py或xsd2pos.py。
- Material Studio脚本xsd2pos.pl。
- OpenBabel。
那如果后续想用其他软件处理优化好的结构呢?那又要去寻找对应的软件。这个时候,ASE的优势就凸显出来了。
在命令行输入命令 ase convert input output ,其中input 为建好的结构文件,output是转化后的文件。
命令行操作
只需要在终端输入下面这行即可将Cu.cif文件转化为POSCAR文件。
ase convert Cu.cif POSCAR
之前大师兄网站讲ASE[1]的时候使用的是这个命令,
ase gui Cu.cif -o POSCAR
试了一下也可以正常转化,不过会有一个UserWarning: You should be using “ase convert …” instead! 的警告。
看来ASE官方还是推荐使用ase convert,直译过来就是ase 转化,需要敲的命令更少,也更好记。
图形界面操作
很多时候建完模希望先打开看一眼结构对不对,再转换成输入文件。可以先用
ase gui Cu.cif
可视化,再通过菜单栏的 File-save 另存为POSCAR。不过这种操作和使用VESTA转化也没什么区别了……
图形界面的好处是可以固定原子!
在图形界面选择原子并固定,另存为POSCAR的时候会把固定的信息也存储下来,即POSCAR中的T T T/ F F F,比使用其他脚本根据z坐标设置固定范围方便得多,也不会固定错原子(血泪教训)。
Python代码操作[2]
本质上就是文件的读和写。
from ase.io import read, writeCu = read('Cu.cif') # 有多帧结构时可以指定index# 一般来说中间会对Cu结构进行一些操作……write('POSCAR', Cu) # write可以有很多参数,不过这样就可以转化为想要的POSCAR文件了
支持的文件转化格式
https://wiki.fysik.dtu.dk/ase/ase/io/io.html#module-ase.io
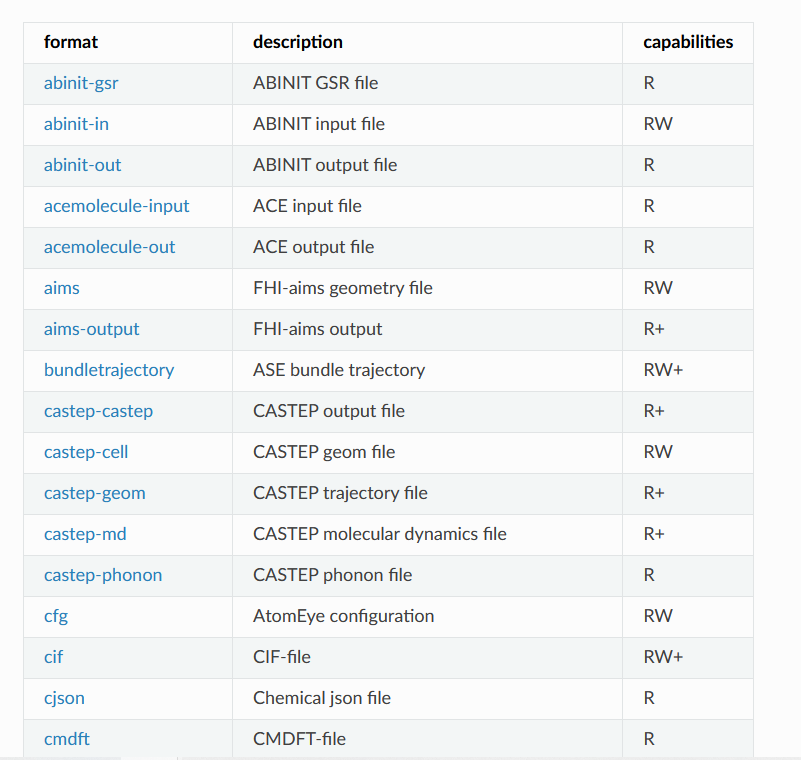
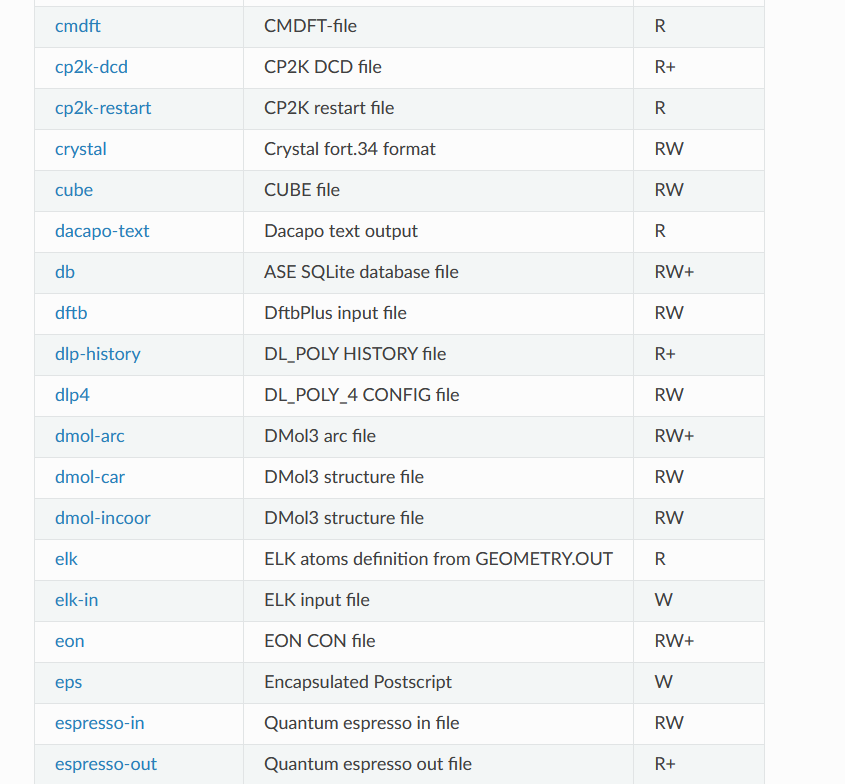
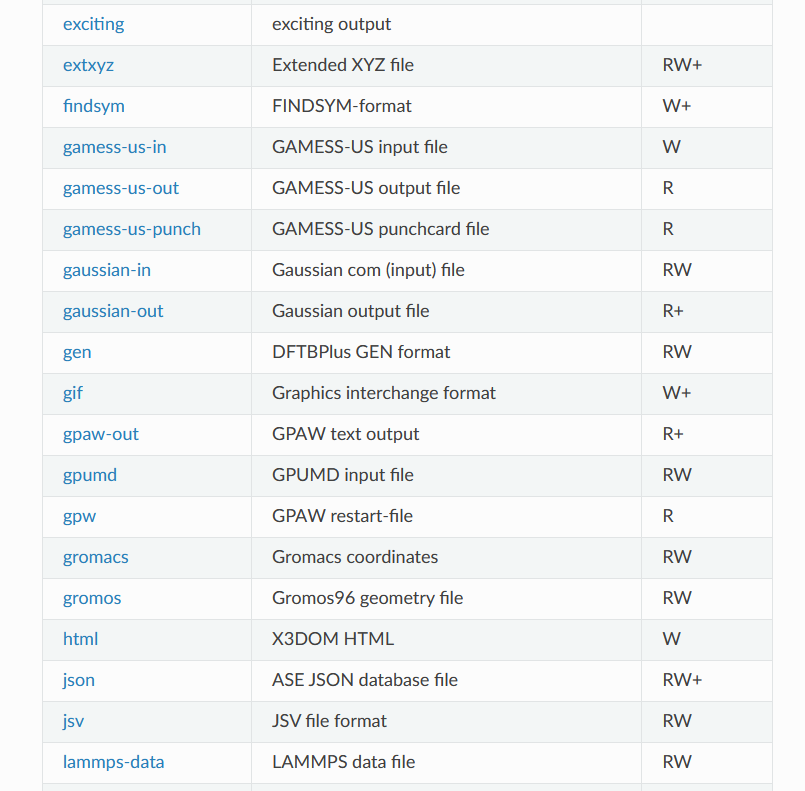

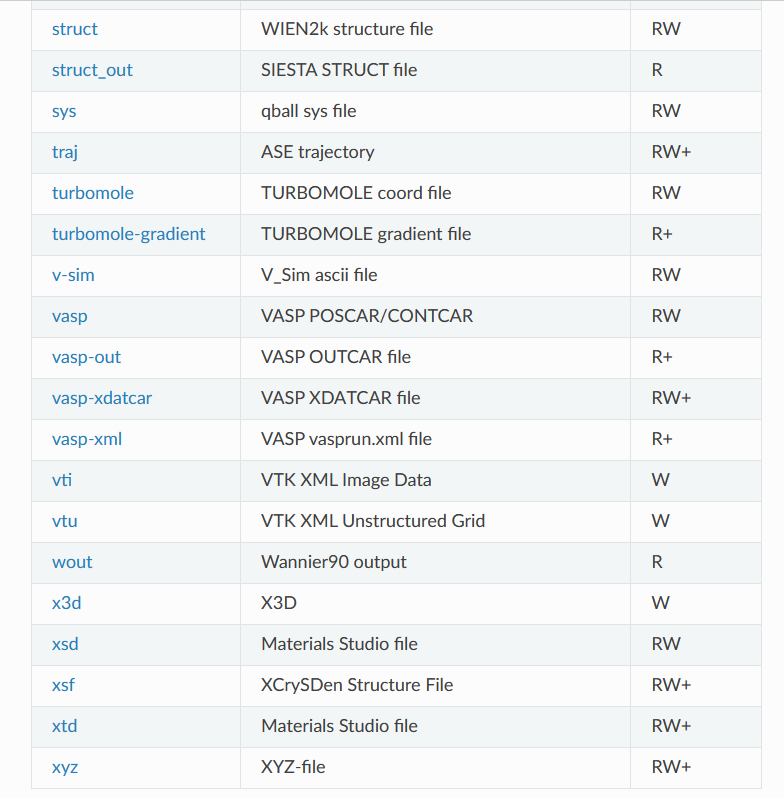
当然,这种文件转化不止适用于POSCAR和cif文件,ASE支持很多很多文件格式,常用的计算软件的输入输出文件都可以进行处理。
https://zhuanlan.zhihu.com/p/585635093?utm_id=0
ASE(三):图形界面查看/建立/修改结构
图形界面召唤
在命令行输入:
ase gui
即可。图形界面长这样
看上去比较普通,但还挺好用的。
查看结构
ase gui 结构名
python代码查看/建立结构
https://wiki.fysik.dtu.dk/ase/ase/build/build.html#module-ase.build
上一篇介绍了怎么用图形界面查看/建立/修改结构,但ASE更常见的应用是基于python代码来研究结构。建议将这篇和上一篇对比着看。
读写、查看结构
from ase.visualize import view #从ASE查看结构的模块visualize导入view
from ase.io import read #导入ASE读取文件的函数readstructure = read('***') #***是结构文件名,支持绝大多数格式
view(structure) #会弹出和命令行使用ase gui ***一样的图形界面#也可以将结构写入文件
from ase.io import write
write('***',structure)
view()函数也可以有其他的关键词,比如view(atoms, viewer=‘VMD’, data=…) 。
- viewer可以有: ‘ase.gui’, ‘gopenmol’, ‘vmd’, ‘rasmol’, ‘paraview’, ‘ngl’, ‘x3d’
- data可以显示3D数据,比如电荷密度。
建立结构
无论是分子还是周期性体系,都可以通过ASE构建。
分子
ASE有一个叫G2的数据库,里面有常见的分子。
from ase.collections import g2
print(g2.names) '''
#数据库里的分子['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF','CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2','C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3','SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane','LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2','methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl','CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN','cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO','trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2','C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C','H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S','N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d','CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O','CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF','C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4','SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O','F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO','C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d','CH3CH2Cl', 'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6','HCOOCH3', 'O', 'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2','CH2SCH2', 'C2Cl4', 'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH','H2CCO', 'CH3CH2NH2', 'Li', 'N2', 'Cl2', 'H2O2', 'Na2', 'BeH','C3H4_C2v', 'NO2']'''from ase.build.molecule import extra
print(extra.keys()) #输出:dict_keys(['Be2', 'C7NH5', 'BDA', 'biphenyl', 'C60'])
使用molecule()函数构建分子:
from ase.build import molecule
atoms = molecule('H2O')
周期性体系
- 纳米管
ase.build的nanotube()构建,默认是碳纳米管,n和m是纳米管的(n,m)标记。
ase.build.nanotube(n,m,length=1,bond=1.42,symbol='C',verbose=False,vacuum=None)
- 石墨烯纳米带
ase.build.graphene_nanoribbon(n,m,type='zigzag',saturated=False,C_H=1.09,C_C=1.42,vacuum=None,magnetic=False,initial_mag=1.12,sheet=False,main_element='C',saturate_element='H')
- 体相
简单的bulk金属可以使用ase.build模块的bulk()函数构建。
ase.build.bulk(name:str,crystalstructure:Optional[str]=None,a:Optional[float]=None,b:Optional[float]=None,c:Optional[float]=None,*,alpha:Optional[float]=None,covera:Optional[float]=None,u:Optional[float]=None,orthorhombic:bool=False,cubic:bool=False,basis=None)
from ase.build import bulk
a1 = bulk('Cu', 'fcc', a=3.6) # 得到Cu原胞,1个原子
a1.cell #查看晶格常数
'''
晶胞:
array([[ 0. , 1.8, 1.8],[ 1.8, 0. , 1.8],[ 1.8, 1.8, 0. ]])
'''a2 = bulk('Cu', 'fcc', a=3.6, orthorhombic=True) #ASE默认,2个Cu原子
'''
array([[ 0. , 1.8, 1.8],[ 1.8, 0. , 1.8],[ 1.8, 1.8, 0. ]])
'''a3 = bulk('Cu', 'fcc', a=3.6, cubic=True) #惯用晶胞conventional cell,4个原子
'''
array([[ 3.6, 0. , 0. ],[ 0. , 3.6, 0. ],[ 0. , 0. , 3.6]])
'''
- 表面
ase.build模块下有一些常见晶面、晶体结构的函数,可以直接调用:
- fcc: fcc100(), fcc110(), fcc111(), fcc211(), fcc111_root()
- bcc: bcc100(), bcc110(), bcc111() * - bcc111_root()
- hcp: hcp0001(), hcp10m10(), hcp0001_root()
- diamond: diamond100(), diamond111()
也可以使用surface()函数构建不那么常见的界面
ase.build.surface(lattice,indices,layers,vacuum=None,tol=1e-10,periodic=False)
比如:
s1 = surface('Au', (2, 1, 1), 9) #Au211面,9层
s1.center(vacuum=10, axis=2) #在c方向加10Å真空层
ASE的局限性在于只能构建简单、常见的结构,在建模效率上可能不如Material Studio点几下就行那么方便,研究复杂结构更多地还是从Materials Project等数据库下载,或者根据文献里的结构进行修改。那这个究竟有什么用呢?个人觉得Python代码的好处是它的灵活性,在需要批量生成结构、与其他代码联用等场景则非常有用;另一个优点则是稳定性,对一个初学者来说,在可视化建模软件点错了几个设置可能会影响建出的模型,但使用同一份Python代码建出来的则是一样的结构。
(使用ASE修改结构后续再慢慢填坑吧。)


)



